
by Marion Duval On my very first day in Sydney, I stopped dead in my tracks to marvel at a flock of large, striking white birds with long, curved black beaks. To my American eyes, they looked exotic and out of place in a small park. A quick Google search Continue Reading
admin
Why should we care about koalas?

by Lily Munro Koalas are one of Australia’s most unique species. Their cute and cuddly appearance has captured the hearts of many tourists and locals alike. Since being listed as Endangered in Queensland, New South Wales and the Australian Capital Territory in 2022, they have also become one of the Continue Reading
Baby’s First Big Conference: SMBE 2024 in Puerto Vallarta

by Toby Kovacs One of the most exciting (and nerve-wracking) milestones in a PhD is dressing your project up, which is often still a little rough around the edges, for a presentation at an international conference. While I’d previously attended several local Australian meetings, last year I had the first Continue Reading
The mother of monsters chooses love not war

by Adele Gonsalvez In Greek mythology, Echidna is known as the “mother of monsters” and is a feared half-woman/half-serpent. However, her possession of mammalian and reptilian traits may be the only similarity between her and her animal namesake. In fact, even when provided with all the weaponry for chemical warfare, Continue Reading
A Guide for Developing Demo-Genetic Models to Simulate Genetic Rescue
Type: Journal article Reference: Beaman, J.E., Gates, K., Saltré, F., Hogg, C.J., Belov, K., Ashman, K., da Silva, K.B., Beheregaray, L.B. and Bradshaw, C.J.A. (2025), A Guide for Developing Demo-Genetic Models to Simulate Genetic Rescue. Evol Appl, 18: e70092. https://doi.org/10.1111/eva.70092 Abstract Genetic rescue is a conservation management strategy that reduces Continue Reading
Genome-wide diversity and MHC characterisation in a critically endangered freshwater turtle susceptible to disease
Type: Journal Article Reference: Nelson, H.V., Silver, L., Kovacs, T.G.L. et al. Genome-wide diversity and MHC characterisation in a critically endangered freshwater turtle susceptible to disease. Immunogenetics 77, 21 (2025). https://doi.org/10.1007/s00251-025-01378-8 Abstract Small, isolated populations are often vulnerable to increased inbreeding and genetic drift, both of which elevate the risk Continue Reading
Why do men exist?
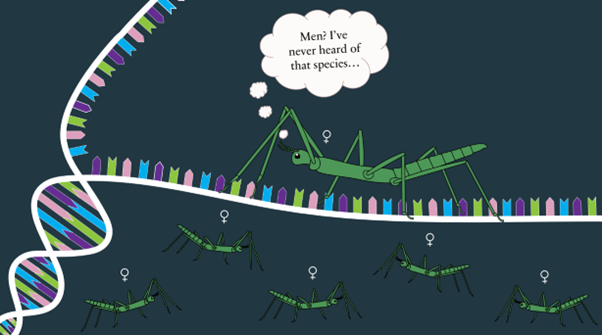
by Soleille Miller This remains one of the oldest unanswered questions in biology, pondered by the earliest philosophers in recorded history. Aristotle explored this very question in his seminal book “On the Generation of Animals”. While his theory (i.e. the male parent contributes the form and the female parent contributes Continue Reading
Range-Wide Assessment of the Tasmanian Devil Gut Microbiome
Type: Journal Article Reference: Molloy, M.M., McLennan, E.A., Fox, S., Belov, K. and Hogg, C.J. (2025), Range-Wide Assessment of the Tasmanian Devil Gut Microbiome. Ecol Evol, 15: e71196. https://doi.org/10.1002/ece3.71196 Abstract The gut microbiome is an important component of host health and function and is influenced by internal and external factors Continue Reading
Marsupial cathelicidins: characterization, antimicrobial activity and evolution in this unique mammalian lineage
Type: Journal Article Reference: Peel Emma , Gonsalvez Adele , Hogg Carolyn J. , Belov Katherine. 2025. Marsupial cathelicidins: characterization, antimicrobial activity and evolution in this unique mammalian lineage. Frontiers in Immunology, 16 – 2025. https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1524092 Abstract Introduction: Cathelicidins are a family of antimicrobial peptides well-known for their antimicrobial and Continue Reading
No Evidence for Distinct Transcriptomic Subgroups of Devil Facial Tumor Disease (DFTD)
Type: Journal article Reference: Petrohilos, C., Peel, E., Batley, K.C., Fox, S., Hogg, C.J. and Belov, K. (2025), No Evidence for Distinct Transcriptomic Subgroups of Devil Facial Tumor Disease (DFTD). Evol Appl, 18: e70091. https://doi.org/10.1111/eva.70091 Abstract Contagious cancers represent one of the least understood types of infections in wildlife. Devil Continue Reading